Genomic Stability
Low copy repeats (LCRs) are typically chromosome-locus-specific sequences sharing greater than 95% sequence identity over a range of 10 to 400 kilobases. These repeats make up at least 5% of the human genome (1). Meiotic recombination between these repeats results in translocation, deletion, duplication and / or inversion of the chromosomal regions flanked by the repeats. Several human diseases are the direct result of this process (reviewed in (2)) including Smith-Magenis syndrome (3), Charcot-Marie-Tooth disease (4), DiGeorge / velocardiofacial syndrome (5) and the inversion form of severe hemophilia A (6, 7). In all disease cases the recombination event between repeats necessarily involved crossing-over. Since homologous recombination between repeated sequences is a major form of mitotic double-strand break repair (8), crossing-over between LCRs mitotically would have the potential for significant genome destabilization.
The evidence that crossing-over can happen as the result
of mitotic repair is much less direct than for meiotic
repair, although a case report of somatic mosaicism in a
female hemophilia inversion carrier is highly suggestive
(9). In order to directly assay mitotic crossing-over as a
double-strand break repair mechanism, I created the XO-GFP
reporter gene (figure 1).
|
|
||||||||||||||||||
|
curved arrow: transcription promoter lightning
bolt: I-SceI cleavage
site red arrow: drug resistance gene thick bars: repeated sequences |
||||||||||||||||||
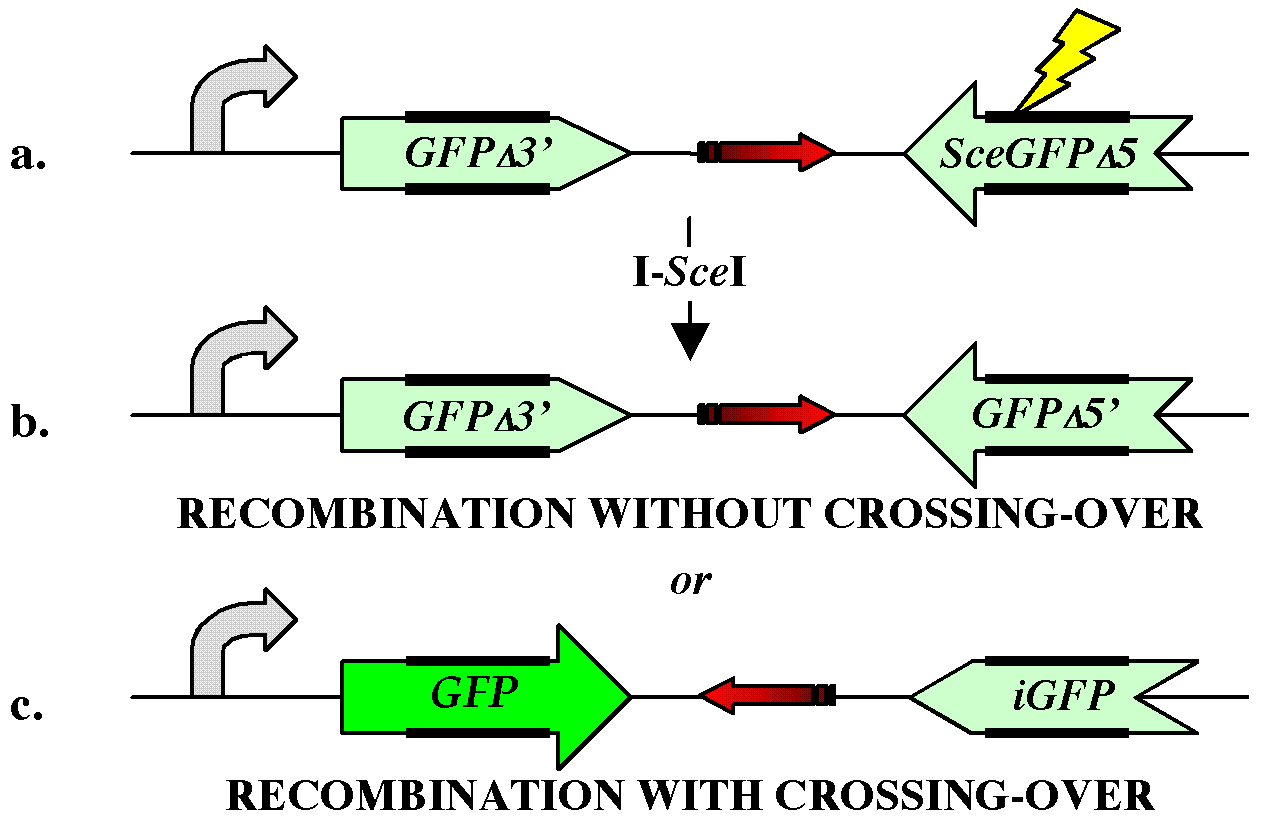
I have integrated the XO-GFP reporter stably into the genome of mouse embryonic stem cells and detected GFP+ cells in response to I-SceI transfection (11). Southern blot analysis of these cells directly demonstrated inversion of the drug resistance marker, confirming mitotic crossing-over in response to a double-strand break. Preliminary evidence indicates that the bias between non-cross-over repair and cross-over repair is on the order of 30 : 1 in favor of non-cross-overs. I will explore the genetic factors responsible for this bias by co-transfecting expression vectors of various DNA repair proteins such as the Bloom's syndrome protein and by reducing expression of these proteins by co-transfection of small inhibitory double-stranded RNA sequences (12).
Of all classes of homologous recombination mediated double-strand break repair, those involving crossing-over have the most dire consequences for genome stability. Crossing-over between either direct or inverted repeats on different chromosomes results in reciprocal translocations. Crossing-over between direct repeats on the same molecule results in deletion of all intervening sequences as an episomal circle, while crossing-over between inverted repeats on the same molecule inverts all intervening sequences as described above. Crossing over between direct repeats on a sister chromatid results in deletions and / or expansions. Finally, crossing over between inverted repeats on sister chromatids leads to the obligate formation of both an acentric and a dicentric chromatid. This latter possibility is especially serious as it can result in prolonged breakage-fusion-bridge cycles (13) which have been identified as an important pathway for the generation of tumor genetic heterogeneity (14). The XO-GFP reporter has the potential to rapidly identify cells in which this process is actively occurring, enabling study of possibly very early events in the cellular progression to cancer.
- E. E. Eichler, Trends Genet 17, 661-9. (2001).
- P. Stankiewicz, J. R. Lupski, Trends Genet 18, 74-82. (2002).
- K. S. Chen et al., Nat Genet 17, 154-63. (1997).
- L. Pentao, C. A. Wise, A. C. Chinault, P. I. Patel, J. R. Lupski, Nat Genet 2, 292-300. (1992).
- L. Edelmann, R. K. Pandita, B. E. Morrow, Am J Hum Genet 64, 1076-86. (1999).
- J. Naylor, A. Brinke, S. Hassock, P. M. Green, F. Giannelli, Hum Mol Genet 2, 1773-8. (1993).
- D. Lakich, H. H. Kazazian, Jr., S. E. Antonarakis, J. Gitschier, Nat Genet 5, 236-41. (1993).
- F. Liang, M. Han, P. J. Romanienko, M. Jasin, Proc Natl Acad Sci U S A 95, 5172-7. (1998).
- J. Oldenburg et al., Blood 96, 2905-6 (2000).
- M. Jasin, Trends Genet 12, 224-8. (1996).
- A. J. Pierce, M. Jasin, unpublished observations.
- S. M. Elbashir et al., Nature 411, 494-8. (2001).
- B. McClintock, Cold Spring Harbor Symp Quant Biol 16, 13-47 (1951).
- D. Gisselsson et al., Proc Natl Acad Sci U S A 97, 5357-62. (2000).